ProBias is a web-server that searches a user-supplied protein sequence for segments that contain unusually dense clusters of user-specified amino acid types and computes analytical estimates of the statistical significance of each cluster. These estimates are based on the discrete scan statistics [1] that allows one to detect sites that exhibit even subtle local deviations from the random independence model. The search is performed using the PBIAS algorithm that addresses the following problem. Given a protein sequence, S, generated using the 20 amino acid types according to the random independence model and a sub-alphabet, B, of m amino acid types (m<<20), find all sub-sequences of S in which residues from B are over-represented. These compositionally biased sub-sequences (segments) will correspond to clusters that have an unusually high density of residues from B (for details see Kuznetsov and Hwang, 2006) (source code and binaries). When PBIAS is used with a sub-alphabet B that contains amino acid types with similar biochemical properties (such as charged, hydrophobic, small, etc) suitable for the identification of a particular type of functional/structural domain or site, in many cases the approximate location of the domain or site in the primary sequence will coincide with the location of compositionally biased sub-sequence(s). For instance, if PBIAS is run with the sub-alphabet of negatively charged amino acids, B={D,E}, then metal binding sites will often be identified (see Fig.1):
Figure 1. A flowchart that shows application of PBIAS to search for
clusters of
negatively
charged amino acids in
a protein sequence.
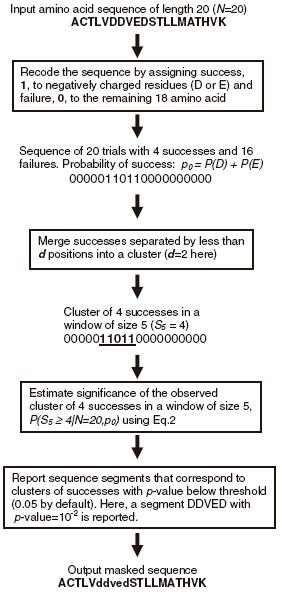
The statistical model used in ProBias takes into account the length of the input sequence. A short compositionally biased segment can be identified as statistically significant in a short input sequence, but the same segment located in a long input sequence may no longer be reported as statistically significant (i.e., it's p-value will be higher than the cut-off value used to report compositionally biased segments). The primary reason for this is that the chance of observing short compositionally biased segments by chance in long sequences is much higher because there is a higher probability of seeing more extreme random fluctuations in amino acid usage simply due to a large number of positions. If you wish to explore long sequences (longer than 300-400 residues), you may want to consider splitting them into domains and analyzing each domain independently.
The user can paste one amino acid sequence in raw format and enter an optional sequence name or upload a sequence file in FASTA format. Maximum allowed sequence length is 1,000 residues.
A FASTA file consists of a header line that begins with a ">" character, followed by an optional sequence name and the sequence itself:
>Sequence name goes here
MALTNAQILAVIDSWEETVGQFPVITHHVPLGGGLQGTLHCYEIPLAAPYGVGFAKNGPT
RWQYKRTINQVVHRWGSHTVPFLLEPDNINGKTCTASHLCHNTRCHNPLHLCWESLDDNK
GRNWCPGPNGGCVHAVVCLRQGPLYGPGATVAGPQQRGSHFVV
twenty characters for twenty amino acid types:A Alanine M Methionine C Cysteine N Asparagine D Aspartate P Proline E Glutamate Q Glutamine F Phenylalanine R Arginine G Glycine S Serine H Histidine T Threonine I Isoleucine V Valine K Lysine W Tryptophan L Leucine Y Tyrosineand the following three characters:
B Aspartate or Asparagine Z Glutamate or Glutamine X UnknownThese one-letter codes can be in either upper-case or lower-case.
SPROT frequencies: use a background model based on the amino acid frequencies observed in the SwissProt database.
PDB frequencies: use a background model based on the amino acid frequencies observed in the Protein Databank. PDB frequencies serve as the background model derived from mostly globular proteins, whereas SPROT frequencies serve as the background model derived from all proteins in the protein universe. The former is better suitable for searching for non-globular domains (such as disordered regions).
Estimate frequencies: use a background model based on the amino acid frequencies observed in the input sequence. This option is used to remove the effect of global compositional bias on local clusters in the case when the input sequence has very unusual amino acid composition.
Linkage distance (integer): the distance used to merge positions into clusters (see Fig.1 for an example). Using smaller values leads to smaller clusters (short biased segments), whereas larger values lead to larger clusters (longer biased segments).
Estimate linkage distance: the linkage distance will be estimated as 2/(3*p{B}), where p{B} is the probability of observing a residue from sub-alphabet B.
P-value cut-off (a decimal number between 0 and 1): the p-value used to identify statistically significant clusters. The web-server will only report clusters with p-value less than this cut-off.
Pre-selected sub-alphabets: the server will use 10 pre-set amino acid sub-alphabets to run PBIAS (each sub-alphabet is used independently). The user will be able to review these 10 sub-alphabets after clicking 'Submit form' button. Please note that the 10 pre-selected alphabets displayed after 'Submit form' is clicked cannot be edited. If you wish to enter your own sub-alphabet(s), check 'User-specified sub-alphabets' option described below.
The ten pre-selected sub-alphabets are as follows:
1. Amino acids with high propensity for helical conformation: ARQEKM.
2. Amino acids with high propensity for beta-sheet conformation: IFTWYV.
3. Amino acids with high propensity for coil (irregular) conformation: NDGHPSTY.
4.Amino acids with high disorder propensity: AHSKDQEG.
Disorder here is defined as protein positions missing electron density in PDB files (so-called Remark465).
Propensities were obtained from the GlobPlot web-server (http://globplot.embl.de/html/propensities.html).
5. Amino acids with high backbone flexibility (wide range of allowed dihedral angles): GHND.
Propensities were obtained from (Kuznetsov and Rackovsky, 2003).
6. Major hydrophobic amino acids: LMFWIV.
7. Major hydrophilic amino acids: RKNDQSTE.
8. Charged amino acids: KRDE.
9. Small amino acids: ANDPSTG.
10. Large amino acids: RFWY.
User-specified sub-alphabets: this option allows the user to specify up to 10 sub-alphabets. The number of sub-alphabets is selected using the pull-down menu. After clicking 'Submit form' button, the user will be prompted to input a name and amino acid characters for each sub-alphabet (only 20 standard amino acid characters are allowed).
The main output page consists of two parts (Fig.2). First part shows a table with the summary of the analysis. Table column 'N' shows the total number of residues from the corresponding sub-alphabet found in the input sequence. Column 'Type' shows whether the residues from this sub-alphabet are over-represented (+) or under-represented (-) in the input sequence. The columns 'Exact p-value' and 'Approx. p-value' show how significant this over- or under-representation is. These columns provide information about global compositional bias of the input sequence. An approximate p-value should be used when the exact p-value cannot be computed. If any statistically significant biased segments were found, the details can be obtained by clicking the link in 'See details' column (Fig.3).
Second part of the main output page shows the input sequence with statistically significant segments printed below the sequence.
The first line below the input sequence shows low complexity regions (if any) found using the PSEG program [2]. PSEG is run with the default arguments. The lines below marked 'Group1', 'Group2',..., show biased segments identified using sub-alphabet 1, sub-alphabet 2,..., etc. In the example shown in Fig.2 PBIAS identifies two different types of compositionally biased segments:
(1) A single segment (line for Group1, cyan color) composed of residues with high coil propensity that corresponds to the exact location of the disordered N-terminal domain of the human prion protein.
(2) A single segment (line for Group2, black color) composed of hydrophobic residues that corresponds to the exact location of the C-terminal signal peptide.
Figure 2. An example of the main output page for the human prion protein.

Figure 3. The details of biased hydrophobic segments found in the human prion protein.
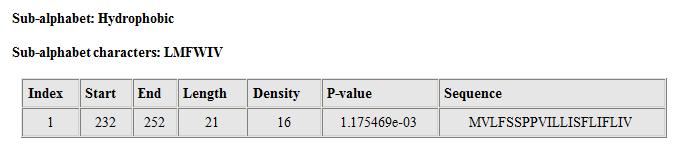
On the details page (Fig.3) all biased segments with p-value less than the cut-off are reported in a table. All segments are numbered in the 'Index' column. Columns 'Start', 'End', and 'Length' show the start and end positions in the input sequence, and length of the segment. Column 'Density' shows the total number of residues from the corresponding sub-alphabet found in the segment. Column 'P-value' shows the p-value of this density. Column 'Sequence' shows the segment itself.
There is no optimal set of parameters that will allow you to find all compositionally biased segments in a single run. The choice of linkage distance is the major factor that affects the results of a search. Smaller linkage distance tends to reveal smaller clusters (shorter compositionally biased segments), whereas larger linkage distance tends to reveal larger clusters (longer compositionally biased segments). We recommend the following simple strategy to optimize you search:
- Try to use relatively short sequences (up to 400 amino acids). If your sequence is longer, consider splitting it into domains. In very long sequences, short compositionally biased segments will not be reported as statistically significant.
- Do not limit your search to the default value of the linkage distance. Try to use different values of the linkage distance (from 1 to 5). Smaller values tend to reveal short compositionally biased segments with high density of amino acids from the target sub-alphabet, whereas larger values tend to reveal longer compositionally biased segments with lower density of amino acids from the target sub-alphabet.
- Finally, try to use 'Estimate linkage distance' option. It will adjust the linkage distance to reflect the makeup of each target sub-alphabet individually.
1. Glaz, J., Naus, J., Wallenstein, S. (2001) Scan Statistics. Springer-Verlag, New York, pp.45-46.
2. Wootton, J.C. and Federhen, S. (1996) Analysis of compositionally biased regions in sequence databases. Methods Enzymol., 266:554-571.
Please address your questions and comments to Igor Kuznetsov